引物的設計和選擇符合熒光PCR的探針并進行設計對于實時熒光PCR尤其重要。可以說,不合理的設計意味著絕對的失敗。但是,好的設計并不等于好的實驗結果,影響PCR和熒光PCR的因素非常多,下面擇其重要進行介紹。
3.1 引物退火溫度
引物的一個重要參數是熔解溫度(Tm)。這是當50%的引物和互補序列表現為雙鏈DNA分子時的溫度。Tm對于設定PCR退火溫度是必需的。在理想狀態下,退火溫度足夠低,以保證引物同目的序列有效退火,同時還要足夠高,以減少非特異性結合。合理的退火溫度從55℃到70℃。退火溫度一般設定比引物的Tm低5℃。
設定Tm有幾種公式。確定引物Tm最可信的方法是近鄰分析法。大部分計算機程序使用近鄰分析法——從序列一級結構和相鄰堿基的特性預測引物的雜交穩定性。所有oligo軟件會自動計算引物的Tm值。在設置退火溫度時可以如下進行:以低于估算的Tm5℃作為起始的退火溫度,以2℃為增量,逐步提高退火溫度。較高的退火溫度會減少引物二聚體和非特異性產物的形成。為獲得最佳結果,兩個引物應具有近似的Tm值。引物對的Tm差異如果超過5℃,就會由于在循環中使用較低的退火溫度而表現出明顯的錯誤起始。如果兩個引物Tm不同,將退火溫度設定為比最低的Tm低5℃。或者為了提高特異性,可以在根據較高Tm設計的退火溫度先進行5個循環,然后再根據較低Tm設計的退火溫度進行剩余的循環。這使得在較為嚴謹的條件下可以獲得目的模板的部分拷貝。
3.2 引物濃度
引物的濃度會影響特異性。最佳的引物濃度一般在0.1到0.5μM。較高的引物濃度會導致非特異性產物擴增。為了確定引物濃度,可以在260nm(OD260)測量光密度值。然后使用光吸收值和微摩消光系數的倒數(nmol/OD),通過Beers法則(公式1)計算引物濃度。
微摩消光系數可以使用公式2計算。與大分子雙鏈DNA可以使用平均消光系數不同,確定引物的精確濃度必須使用計算的消光系數。這是因為引物較短,堿基組成差異很大。在oligo軟件上可以計算出引物的的消光系數(OD/μmol)和消光系數的倒數(μmol/OD)。
一般商業合成的引物以O.D.值表示量的多少,在一般情況下,20個堿基長的引物,1個O.D.加100ul水后引物濃度為50pmol/ul(50μM)。也可以用OLIGO軟件,根據引物的的消光系數(OD/μmol),計算出一定的工作濃度下,引物的加水量。
濃度(μM)=A260(OD/ml)×稀釋系數×消光系數的倒數(nmol/OD)
舉例:計算某寡核苷酸(溶于1ml水中),取其中的10μl稀釋100倍(加入990μl)水中。在A260處測吸光度為0.2。計算消光系數的倒數為4.8 nmol/OD,代入得:
濃度=0.2(OD/ml)×100×4.8(nmol/OD)=96nmol/ml=96μM
3.3 引物、探針的純度和穩定性
定制引物的標準純度對于大多數PCR應用是足夠的。部分應用需要純化,以除去在合成過程中的任何非全長序列。這些截斷序列的產生是因為DNA合成化學的效率不是100%。這是個循環過程,在每個堿基加入時使用重復化學反應,使DNA從3'到5'合成。在任何一個循環都可能失敗。較長的引物,尤其是大于50個堿基,截斷序列的比例很大,可能需要純化。
引物產量受合成化學的效率及純化方法的影響。定制引物以干粉形式運輸。最好在TE重溶引物,使其最終濃度為100μM。也可以用雙蒸水溶解。
引物的穩定性依賴于儲存條件。應將干粉和溶解的引物儲存在-20℃。以大于10μM濃度溶于TE的引物在-20℃可以穩定保存6個月,但在室溫(15℃到30℃)僅能保存不到1周。干粉引物可以在-20℃保存至少1年,在室溫(15℃到30℃)最多可以保存2個月。
探針即寡核苷酸進行熒光基團的標記,標記本身有效率的區別。探針標記以后一般應該純化,純度高、標記效率高的探針不僅熒光值高,且保存時間可以高達一年以上。不同的生物技術公司探針標記效率和純度有很大的區別。
由于反復凍融易導致探針降解,在確認探針質量好的情況下,最好稀釋成2uM(10×),作為工作濃度分裝多支,避光保存。
3.4 熱啟動
熱啟動PCR是除了好的引物設計之外,提高PCR特異性最重要的方法之一。盡管Taq DNA聚合酶的最佳延伸溫度在72℃,聚合酶在室溫仍然有活性。因此,在進行PCR反應配制過程中,以及在熱循環剛開始,保溫溫度低于退火溫度時會產生非特異性的產物。這些非特異性產物一旦形成,就會被有效擴增。在用于引物設計的位點因為遺傳元件的定位而受限時,如定點突變、表達克隆或用于DNA工程的遺傳元件的構建和操作,熱啟動PCR尤為有效。
限制Taq DNA聚合酶活性的常用方法是在冰上配制PCR反應液,并將其置于預熱的熒光定量PCR儀。這種方法簡單便宜,但并不能完全抑制酶的活性,因此并不能完全消除非特異性產物的擴增。
熱啟動通過抑制一種基本成分延遲DNA合成,直到PCR儀達到變性溫度。包括延緩加入Taq DNA聚合酶在內的大部分手工熱啟動方法十分煩瑣,尤其是對高通量應用。其他的熱啟動方法使用蠟防護層將一種基本成分,如鎂離子或酶,包裹起來,或者將反應成分,如模板和緩沖液,物理地隔離開。在熱循環時,因蠟熔化而把各種成分釋放出來并混合在一起。象手動熱啟動方法一樣,蠟防護層法比較煩瑣,易于污染,不適用于于高通量應用。
Transitor? Hot Start Taq酶對于自動熱啟動PCR來說高效可靠。Transitor? Hot Start Taq是在常規Taq酶的基礎上進行了化學修飾,使得該酶在低溫或常溫下沒有酶活,因此在加樣至PCR擴增前,不會產生由于引物隨機粘連而形成的非特異性擴增。高溫條件下,化學修飾集團會被剪切,從而釋放酶活。此外,隨著PCR擴增的進行,酶活會被逐步釋放,有效提高擴增效率及產物量。Transitor? Hot Start Taq DNA聚合酶在變性步驟的95℃保溫過程中要持續20分鐘才會被釋放到反應中,從而完全恢復聚合酶活性。
3.5鎂離子濃度
鎂離子影響PCR的多個方面,如DNA聚合酶的活性,這會影響產量;再如引物退火,這會影響特異性。dNTP和模板同鎂離子結合,降低了酶活性所需要的游離鎂離子的量。最佳的鎂離子濃度對于不同的引物對和模板都不同,但是包含200μM dNTP的實時定量PCR,使用3到5mM帶有熒光探針的鎂離子溶液(普通PCR是1.5mM)。較高的游離鎂離子濃度可以增加產量,但也會增加非特異性擴增,降低忠實性。為了確定最佳濃度,普通PCR從1mM到3mM,以0.5mM遞增,進行鎂離子滴定。為減少對鎂離子優化的依賴,可以使用 Transitor? Hot Start Taq酶。Transitor? Hot Start Taq DNA聚合酶能夠在比一般的Taq DNA聚合酶更廣的鎂離子濃度范圍內保持功能,因此僅需較少的優化。
3.6 模板質量
模板的質量會影響產量。DNA樣品中發現有多種污染物會抑制PCR。一些在標準基因組DNA制備中使用的試劑,如SDS,在濃度低至0.01%時就會抑制擴增反應。分離基因組DNA較新的方法包括了DNAZol,一種胍去垢劑裂解液,以及FTA Gene Guard System,其可以同基質結合,在血液及其他生物樣品中純化貯存DNA。應注意進行PCR 反應的模板質量,以增加PCR 反應的成功率。
3.7 模板濃度
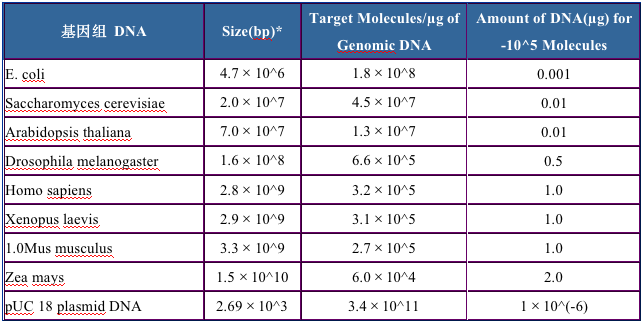
起始模板的量對于獲得高產量很重要。對大多數PCR擴增和熒光PCR擴增,104到106個起始目的分子就足以觀測到好的熒光曲線(或在溴化乙錠染色膠上觀察到)。所需的最佳模板量取決于基因組的大小(下表)。舉例說,100ng到1μg的人類基因組DNA,相當于3×104到3×105個分子,足以檢測到單拷貝基因的PCR產物。質粒DNA比較小,因此加入到PCR中的DNA的量是pg級的。對于一般的檢測樣品,10——100ng的量就足夠檢測了。當然對模板做一個梯度稀釋,對于定量PCR而言是非常容易,且能分析擴增效率。
表1. 基因組大小和分子數目的比例
基因組大小和分子數目的比例
3.8 防止殘余(Carry-over)污染
PCR易受污染的影響,因為它是一種敏感的擴增技術。小量的外源DNA污染可以與目的模板一塊被擴增。當前一次擴增產物用來進行新的擴增反應時,會發生共同來源的污染。這稱之為殘余污染。從其他樣品中純化的DNA或克隆的DNA也會是污染源(非殘余污染)。
可以在PCR過程中使用良好的實驗PCR步驟減少殘余污染。使用帶濾芯的移液管可以阻止氣霧劑進入eppendorf管內。為PCR樣品配制和擴增后分析設計隔離的區域,在準備新反應前更換手套。總是使用不含有模板的陰性對照檢測污染。
使用預先混合的反應成分,而不是每個反應的每個試劑單獨加入。
一種防止殘余污染的方法是使用尿嘧啶DNA糖基化酶(UDG)。這種酶(也稱為尿嘧啶-N-糖基化酶或UNG)移除DNA中的尿嘧啶。在擴增過程中將脫氧尿嘧啶替換為胸腺嘧啶使得可以把前面的擴增產物同模板DNA區分開來。因為前面的擴增產物對UDG敏感,所有可以在PCR前對新配制的反應用UDG處理以破壞殘余產物。
![雙[三(羥甲基)氨基甲烷],CAS:6976-37-0](http://struc.chem960.com/strucimg/7000/ctzw0nzi34oyob9fnlmhiwee.png)